New anti-COVID drug candidates


Around the time that delta was emerging and the trials of one of the now-accepted vaccines were being run in Oxford, I read a really interesting account (in New Scientist) on how the test subjects were chosen, the conditions under which they had to agree to live while remaining part of the trials, how this then affected the rest of their lives as the UK (or other countries, depending on which part of the consortium the subjects lived in, and how high the Community contagion factor was there) etc.
Truly fascinating, particularly when you think of such things as who’s got the placebo? who’s in the control group? If they’re all living in the community, then how do you control every possible variable in daily life? etc. The interviews were wonderful reading, as the subjects’ motivations and emotions were also revealed - test conditions prevented them from participating in some community events as lockdowns were lifted (because they weren’t double-jabbed by a set date, for example) etc.
As I’ve written elsewhere over the years, I’ve got a stack of allergies and sensitivities that we now know to be due to mast cell activation disorder (which isn’t IgE mediated so testing for it is really hard). My biggest challenges with these vaccines are the base fluids, colours and the preservatives - things often seen considered almost inert in the bigger scheme of things. So in many ways a great potential test subject, yet also a nightmare one since a loud noise or a nasty surprise could set off a bad reaction that my overactive immune system will associate with the vaccine.
(I’ve also got thunderstorm asthma, and Crohn’s, so I’m averaging a covid test every 4-6 weeks at present as cyclone season and La Niña build up) I think I’m dreading Omicron more than any other version of covid, so far even though I’m double-vaxxed.

Getting back to the general topic here, I want to discuss some things about selective toxicity.
Penicillin has a small risk of toxicity. Compared to other biologically active substances, clinicians can administer these drugs at relatively high doses without harming patients. Estimates are that it would take 5g/kg body weight intravenously to cause convulsions in a patient.
Five grams per kilogram??? Holy crap, that would mean a dose of around 350 GRAMS for your typical 70 kg man to cause seizures. More than half a pound? Okay, I think it'll be safe when we give an IV dose of 3 grams. Okay, enough silliness but the thing to note is that with selective toxicity, substantial doses of an antibiotic that will overwhelm a susceptible bacteria are possible because we don't need the things they **** with.
Selective toxicity is relatively easy when it comes to killing prokaryotic (like bacteria & others) organisms versus eukaryotes (almost everything else, including mammals). Prokaryotes like bacteria have cell walls. Since they are dividing at such a high rate (every 20 minutes or so), the cell wall is constantly being remodeled, mainly by linking up and unlinking peptidoglycan molecules. They're like bricks in the walls of a house that is constantly being renovated. What's nice is ---- mammals have cell membranes but don't have cell walls around them like bacteria do.
So, if we can find something that messes up cell walls and not much else, maybe we got something here. We weren't having much luck at the turn of the 20th century and we can thank Alexander Fleming for being a little sloppy one day with his bacterial cultures for the start of it all. One of them got contaminated with a Penicillium fungus and before he threw it out he noticed that the areas around the mold spots did not have any bacteria in them. Hmmm? How come? Eventually, they discovered that some chemical secreted by that fungus messed up the enzyme that manipulates the peptidoglycan molecule. No cell wall maintenance = death for the bacteria he was studying. Those chemicals were named penicillins and lettered A, B, .... and penicillin G turned out to be the best one to use as a drug. It had to be given as an IV or IM (ouch!!!) injection though since it was destroyed in the GI tract if you took it by mouth. Penicillin V worked best as an orally active drug.
In addition to the penicillins and cephalosporins, the other classes of antibiotics we have today all have a similar selectively toxic effect on bacterial, only targeting various other differences between prokaryotes and eukaryotes. A big difference is in the ribosomes bacteria use compared to ours. There are several classes of antibiotics that mess up bacterial protein synthesis as a way of killing them or at least stunting their growth. In certain serious infections, antibiotics are doubled or even tripled up in a patient to attack the invading bacteria at multiple vulnerable points.
More on this later but viruses are very different from mammalian cells but they're harder to kill than bacteria because they use OUR OWN enzymes to do some of the things needed to reproduce. Those bastards.

Before departing from bacteria, I wanted to mention resistance to things that kill them. Since they divide so quickly, the odds of some mistake in replicating their DNA changing something enough to evade an antibiotic are high. It almost seems like bacteria have brains and can learn from what is going on a come up with some weapon to protect themselves with. A slightly modified protein that the antibiotic does not bind is one thing but certain bacteria came up with an enzyme, ß-lactamase, that actually breaks down penicillin molecules by cleaving its ß-lactam bond. Well how the hell did it do that? Just think millions of years times 20-minute generations and trillions and trillions of bacteria trying to survive attacks by fungal organisms and it isn't surprising.
Almost every different bacterial species has some way of defending itself against chemical attacks, including things that they didn't have eons to learn about. To make matters worse, bacteria have ways of "sharing" these resistance genes by way of plasmids, independent bundles of genetic material that are able to freely float in the surrounding fluids and get taken in by neighboring bacteria. Since they have similar enzymes, it isn't like there needs to be some sort of currency exchange and there's a high chance of different species being able to work with these plasmids. Thus, a bacteria that has never encountered a given antibiotic can still be resistant to it anyway through pure luck.
In our bodies, there are so many different bacterial members of our normal flora so it's definitely possible and perhaps a dang miracle that we are still here on earth. We can tolerate these bacteria and even benefit from them in symbiotic ways as long as they remain where it's safe for them to be. Most of them are on our skin and in our digestive tract and lord help the person who ever has a perforation in their large intestine that lets these bacteria out into the rest of the body. Appendicitis that isn't caught in time, anyone? At least now we have reliable surgical techniques and broad-spectrum antibiotics to save patients who don't get to an ER fast enough.
Peter, I can’t remember who did the research or where but I remember reading (again in New Scientist I think referring to a PLoS paper) about a family of single-celled organisms that successfully invade their hosts by acting as a coordinated group of cells at critical times. The study looked at the life cycle as well as how they communicate.
Again, fascinating.
Here's a sort of analogy about antibiotic resistance. Early last month, some friends of mine raced IronMan Texas and some riders had unfortunate incidents on the bike portion of the triathlon because someone deliberately scattered lots of sharp objects across the road at one point. Most of them were lucky and didn't run over one but lots of racers did. Howard was one of them but because the race organizers found out about it, they sent support techs out onto the course to help riders get back on the road faster so they weren't t too much of a disadvantage. Howard didn't need the help though. He's good at this stuff and was rolling again pretty quickly. I think he said it was about 7-8 minutes for him once he realized he'd flatted. He qualified for Kona anyway, which is a big thing if you know anything about IronMan tris.
Those small things were selectively harmful to cyclists because their tires are smaller and much thinner than car & truck tires.
Well, what can cyclists do? They could use tires that have thicker walls but that doesn't work for competitive cyclists because the heavier rotating weight makes a wheel harder to turn and get rolling. Sure, it could maintain greater momentum once it's going and that would be great, but what about working against gravity up a long climb? Okay, so what else? You could use a protective liner between the tire and inner tube - nah. They've been making tubeless tires for years now and they are used with a sealant that is infused inside to quickly seal up any punctures. It works pretty good but there are trade-offs there, too.
https://www.triathlete.com/gear/bike/ask-chris-what-are-the-best-tires-for-triathletes/
Those tires that are less likely to puncture are analogous to the different ways bacteria can have resistance to something. Bacteria have their ways and viruses have theirs, too, like random point mutations in their mRNA genome that can cause a different shape for some point on a protein (like the COVID spike protein) that is enough to confer resistance to antibodies that successfully attack the old version. The new protein might still have the ability to do its dirty work but now it isn't recognized like the old one. Hey, it can happen. Most mutations cause dysfunction that kills the virus or makes it harder for it to survive and so those strains do not thrive.
joanne said:
Peter, I can’t remember who did the research or where but I remember reading (again in New Scientist I think referring to a PLoS paper) about a family of single-celled organisms that successfully invade their hosts by acting as a coordinated group of cells at critical times. The study looked at the life cycle as well as how they communicate.
Again, fascinating.
Hey, don't scare us. This is a kids' show for goodness' sake. ;-)
Might have been a host-tree thing, and overall relationship was beneficial (in the dense Amazon? New Guinea?)
(in the dense Amazon? New Guinea?)
PeterWick said:
Hey, don't scare us. This is a kids' show for goodness' sake. ;-)
Really wish I could remember the details.
Pfizer also has an orally active antiviral medicine, called Paxlovid (PF-07321332 + ritonavir), that we hope is effective against COVID. Unlike molnupiravir, which messes with the production and expression of mRNA, PF-07321332 inhibits an important viral protease. Scientists found an enzyme the virus uses to break down a specific protein in some important step in its life cycle and PF-07321332 fucks with it.
EDITED TO ADD: Paxlovid is not just ritonavir as I'd originally written. Ritonavir is added to slow down the clearance of PF-07321332, the actual protease inhibitor.
Do you recall the early anti-HIV drugs like AZT? They messed with the way HIV replicates itself because they are analogs of the building blocks in genetic material. That is somewhat similar to how molnupiravir works. AZT and its cousins were no picnic and they weren't very effective but what else was there? So, they were approved because geez, we were desperate for something, anything. It wasn't until the mid-90s when the protease inhibitors arrived that AIDS treatment became realistic hope. Now there was the ability to use combinations of drugs to have a multipronged attack on HIV. Things have improved but it has been 40 years and we still don't have a definite cure. We can suppress HIV enough to live well though. And drug manufacturers aren't exactly knocking on the FDA's door with any vaccines against HIV, too.
It's kinda the luck of the draw that COVID isn't just like HIV. Can you imagine? 40 years and this many people are still dying each year. And it's not like idiots can **** can blame COVID on drug use and sex.
https://www.unaids.org/en/resources/fact-sheet
- In 2020, around 680 000 [480 000–1 million] people died from AIDS-related illnesses worldwide, compared to 1.9 million [1.3 million–2.7 million] people in 2004 and 1.3 million [910 000–1.9 million] people in 2010.
Pfizer's drug certainly has some promise. The big quote they probably want everyone to remember is:
The scheduled interim analysis showed an 89% reduction in risk of COVID-19-related hospitalization or death from any cause compared to placebo in patients treated within three days of symptom onset (primary endpoint)
They've already conducted studies in three different groups:
Those that are at high-risk of progressing to serious disease after contracting COVID.
Those that are at what they called a standard risk of getting badly ill.
Those that have been exposed because they were a close contact of a COVID+ person. This one is key since it could show that Paxlovid works as a prophylactic treatment if exposure is caught soon enough.
Paxlovid works quite well if a COVID+ person is started on it within 3 or even 5 days after symptoms appear. Plus, they studied a dosing regimen that is very reasonable. Given orally every 12 hours for five days would be great if that's all that most cases require.

https://www.nature.com/articles/s41594-021-00651-0 This drug pulls a neat trick with historical background
Merck's molnupiravir is a pill you can take orally and it's in phase III trials for treating COVID-19. Molnupiravir sharply jacks up the appearance of mutations in the viral RNA as it tries to replicate which messes up its survival in experimental models and now in humans. COVID uses much of our cell's components to make more of itself but it still has to use something that it has that mammals don't. That's what this drug attacks to have a selectively toxic effect on the virus and not us, even though we both use mRNA to live.
How's this for a name? Viral RNA-dependent RNA polymerase - yeesh, no wonder they use the RdRp abbreviation. Molnupiravir gets absorbed from the intestines and then metabolized into its active form, a cytidine imposter named β-D-N4-hydroxycytidine (NHC) triphosphate. The RdRp enzyme gets fooled by it and picks it up when it should be grabbing a cytidine triphosphate or uridine triphosphate molecule. So each time an RNA strand calls for a C or a U to be put into the strand next, this enzyme has a strong chance of grabbing and putting in NHC by mistake. So if you can get NHC into the cells in a high enough concentration you have a good shot at gumming up the works. Since molnupiravir (it may take another week before I can spell it without looking at it) is not picked up easily by the mammalian RNA polymerase enzymes it can be dosed in pretty large amounts. 800 mg twice a day is a big dose. Not many non-antiinfective drugs need such big doses but there are a few common ones. Ibuprofen (600-800 mg) or naproxen (375-500 mg) anyone? Anyway, since it isn't likely to cause the mutations in our mRNA we can dose the hell out of a patient and that's what is needed to kill COVID as fast as possible.
Okay, so that's step one. Using the imitator of C and U residues to cause errors in the "mirror image" RNA strand the virus makes of its genome. All those goofs are point mutations. It's like buckshot spraying all over an area versus single bullets.
Now step 2 is that same viral enzyme having to use the mistake-ridden RNA as a template to make the mRNA it then gets our cells to make the viral proteins from. When the RdRp uses the goofed-up RNA as a template, NHC sitting in the sequence fools it a second time. The RdRp can't tell what it is, a C or a U, and so grabs either adenine (A to match a U) or guanine (G to match a C) and stitches it in. in addition to being mistaken for C and U, the NHC molecule can still form a base pair with either A or G. They think that's why these messed up RNA strands generate heavily mutated RNA. Those many mutations in the mRNA cause nonsensical strands of amino acids to be produced when it is translated instead of functional enzymes.
This mechanism that causes mistakes in two different steps of the process could be the reason that this drug inhibits the growth of many different viruses. NHC is bound by other viral RNA polymerases. It's like a broad-spectrum antibiotic; one that is useful in several cases where a doctor has identified the bug causing the patient's infection but also a good choice for when a patient is in trouble but you don't know what bug they have yet.
A broad-spectrum antibiotic covers many of the usual suspects so doctors start the patient on that to buy them time while they figure out what is wrong. Sometimes two or even three different ones are started to really protect against a lot of pathogens. If the bug they identify is susceptible to a drug you're already using, great. Sometimes they don't even bother to finalize the diagnosis but if you wind up in a hospital there's going to be a lot of culture tests to make sure they're using the best possible choice. This same drug could be used against many RNA viruses that currently bother us.
Sounds great, right? Perhaps. Clinical trials haven't shown it to be really effective on its own. If Pfizer's protease inhibitor drug, Paxlovid, doesn't work well enough as a single agent, these two drugs could be used together. That's similar to the anti-HIV treatment regimens.
But when have you ever known me to be absolutely sure of medical things? It turns out that HIV treatments haven't been good for everyone because of specific properties in a patient's mitochondrial DNA. Mitochondrial DNA is only one chromosome with 37 genes (last time I checked) and there are differences that develop and stabilize in a person. This source of DNA is used for checking matrilineal heritage in greater detail by the way. We're learning more and more about the role of mitochondrial DNA in different kinds of adverse drug reactions or just plain differences in response. Some people just do not tolerate one drug but are fine with another that does the same thing and we've been left to wonder why.
They're not sure what it is about antiviral drugs but weird drug effects are traced to mitochondrial DNA differences. The same sort of thing might happen with molnupiravir or its cousins but one reason to be optimistic is that molnupiravir is only a short-term exposure drug while the current HIV drugs are taken for life.

RobB said:
Paxlovid is already a combination of two drugs.
Yes, I discussed that up above with the first post about it.
From November 27
EDITED TO ADD: Paxlovid is not just ritonavir as I'd originally written. Ritonavir is added to slow down the clearance of PF-07321332, the actual protease inhibitor.
I'd written too soon. The lay press tends to simplify things a bit and I didn't look deeper before doing that post. Paxlovid refers to the product, a combination of the compound named PF-07321332 and ritonavir. Lopinavir/ritonavir works on some viruses but it is not effective against COVID. What it does do in this case is get in the way of enzymes that would clear out PF-07321332 too quickly if it was given alone.
Here is a more common example of the way a product's brand name is considered the drug name but there are multiple drugs together. Ever hear of Augmentin, the commonly used antibiotic? That's the product's brand name but it is actually a combination of amoxicillin and clavulanic acid. Amoxicillin used to be okay on its own but now there are many bacteria that are resistant to it because they have a ß-lactamase enzyme that breaks it down quickly. Clavulanic acid isn't a good antibiotic per se but it wrecks ß-lactamase enzymes permanently because of the way it covalently binds a serine residue in that enzyme's active site. As long as there's enough clavulanic acid present in high enough concentrations, amoxicillin can get to work. The product, Timentin, is based on the same principle. It's a combo of ticarcillin and clavulanic acid.
Why don't they just use a different antibiotic if they don't work? It isn't so simple. Amoxicillin and ticarcillin are still effective against some common bacteria and using them as a first-line agent is worth it so as to save more modern antibiotics for down the road against resistance. Many antibiotics just aren't useful after a while because bacteria develop resistance that isn't as easily countered as with these two products.
Paxlovid (ritonavir & PF-07321332) and molnupiravir might be used in a multidrug approach if it comes to people needing immediate help, especially if they weren't a suitable candidate for vaccination and were left vulnerable.
From the looks of things, if they're approved, either drug or their combination, would be indicated for brief outpatient anti-COVID treatment. Somewhat like Tamiflu but a more modern version for a more serious threat.
I do a lot of searching just because trying to figure things out or better yet, trying to fix/mess with things, is something I may never shake off. Just ask my wife. I'm also not a strong advocate for treating illnesses with drugs if they can be avoided. I prefer the older pursuit of discovering medicines from plant sources, called pharmacognosy, or from microbes or animals. I got hooked on that discipline because my research used more naturally sourced compounds than synthetic ones. Looking for drugs of all kinds in nature still goes on today, and with the new tools scientists have developed, so much more work can be done. It isn't exactly quantum computing but bioinformatics and molecular pharmacology techniques have made the stuff we came up with in the '90s look like neolithic cave remnants.
And so we have the following - and sorry, but the two key molecules, cannabigerolic acid and cannabidiolic acid, come from the hemp plant and you won't get them from marijuana without some complicated chemistry work. See the link at the bottom of this post.
Cannabinoids Block Cellular Entry of SARS-CoV-2 and the Emerging Variants
https://pubs.acs.org/doi/10.1021/acs.jnatprod.1c00946#
As a complement to vaccines, small-molecule therapeutic agents are needed to treat or prevent infections by severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2) and its variants, which cause COVID-19. Affinity selection-mass spectrometry was used for the discovery of botanical ligands to the SARS-CoV-2 spike protein. Cannabinoid acids from hemp (Cannabis sativa) were found to be allosteric as well as orthosteric ligands with micromolar affinity for the spike protein. In follow-up virus neutralization assays, cannabigerolic acid and cannabidiolic acid prevented infection of human epithelial cells by a pseudovirus expressing the SARS-CoV-2 spike protein and prevented entry of live SARS-CoV-2 into cells. Importantly, cannabigerolic acid and cannabidiolic acid were equally effective against the SARS-CoV-2 alpha variant B.1.1.7 and the beta variant B.1.351. Orally bioavailable and with a long history of safe human use, these cannabinoids, isolated or in hemp extracts, have the potential to prevent as well as treat infection by SARS-CoV-2.
----------------------------------
I included the picture representing the spike proteins receptor-binding site because it gives you a sense of the 3-D arena medicinal chemists are "living in" when they are searching for compounds that interact with that site in the way they want. I figure in the last couple of years tens of thousands of compounds, both organic and synthetic, have been examined in a desperate effort at solving COVID. As they were figuring out the piece to put in the vaccines, they also produced just that section of the spike protein in such a way that it is able to fold into its conformation similar to how it is shaped in the whole spike protein. That let them do two types of experiments. They could test compounds to see if they bound that protein in the binding site. Since they also proved that this protein fragment bound the angiotensin-converting enzyme (ACE) on the surface of human epithelial cells, they could also see which of the compounds that bound the spike protein fragment would also block that fragment from binding the ACE on the surface of epithelial cells. Doing that would stop a COVID infection in its tracks.
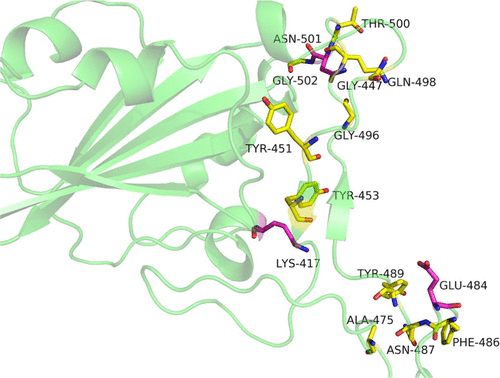
At the end of that article they discussed the prospects for those compounds as COVID treatments. It would take some stiff oral doses but they might be able to achieve a high enough concentration to block effectively COVID virus binding & entry. It might be possible to come up with a nasal spray or an inhaler formulation but I don't think that would work. That said, I haven't been in a pharmaceutical dosage form lab in a long time.
The other big thing they discussed was the notion that those compounds disrupt the interaction of the spike protein with a target that DOESN'T change because of mutations. The ACE enzyme is what the COVID virus uses as its doorway so however it might mutate and escape antibodies against earlier versions of itself, it must still bind the ACEs located on the cells it invades. Those cannabinoids get in the way like someone in a phone booth fishing for that last quarter in their pocket to make a call. And that's something that would work great in combination with vaccines or those two drugs mentioned above.

Sponsored Business





Promote your business here - Businesses get highlighted throughout the site and you can add a deal.
Employment Wanted
Latest Jobs
Employment Wanted
-
Nomadic Notary: Professional Notary Services Available!
Apr 22, 2024 at 3:43pm
-
Apr 22, 2024 at 12:46pm
-
Apr 22, 2024 at 10:49am
-
Apr 20, 2024 at 8:18pm
-
Brazilian cleaning 973 776 2481
Apr 20, 2024 at 4:48pm
Help Wanted
-
CKF600 Part Time Nanny for Toddler (ASAP Start)
Apr 22, 2024 at 3:21pm
-
Full Time Nanny Needed for 2 in Essex County (June/July Start)
Apr 20, 2024 at 8:42pm
-
NPF509 FT Nanny/Family Assistant for Twins (ASAP Flex)
Apr 19, 2024 at 12:38pm
-
CF582 FT Nanny/Family Assistant for 2 (Late May Start)
Apr 19, 2024 at 12:18pm
-
SF5001 FT Nanny for 2 (ASAP Start)
Apr 19, 2024 at 12:02pm
Lessons/Instruction


Advertisement




Alas, Merck and Ridgeback Biotherapeutics had high hopes for this drug but molnupiravir doesn't have the efficacy an earlier trial's data suggested. That first trial had 775 subjects and the latest results are from studying more than 1400 patients. A 50% reduction in hospitalization/serious illness sounded promising but now they've found it is more like a 30% reduction. This doesn't mean the whole approach behind that drug is a failure though. That drug may have a few different reasons why it didn't perform so well.
https://www.nytimes.com/2021/11/26/science/merck-molnupiravir-antiviral-covid-pill.html
Those large numbers are easier to talk about but it is a bit deeper in terms of the real numbers which show a mix of good news and bad depending on how worried one is. This is from the report:
That drug takes it down from a 1 in 10.3 chance of getting really sick to a 1 in 14.7 chance - presenting the numbers that way does look a little odd, especially when the benefit is so small. On the other hand, there's a low percentage of people who are infected that will need hospitalization in the first place. Frankly, it would make me nervous to be in such a trial knowing that some are going to be given a placebo rather than the drug in question. In cases such as these, the inclusion criteria are important.
https://www.merck.com/news/merck-and-ridgeback-biotherapeutics-provide-update-on-results-from-move-out-study-of-molnupiravir-an-investigational-oral-antiviral-medicine-in-at-risk-adults-with-mild-to-moderate-covid-19/
I didn't find a manuscript on the actual trial, only that report on Merck's website. It says "at-risk adults" were the tested population but what that means is unclear since we don't see the methods section that would normally be in a published manuscript. Are they vaccinated or unvaccinated adults? What brand of vaccine? One or two doses? Boosted or not? Only those with other health issues that would make them more susceptible to COVID's harms? And so on. Those details will be available to FDA analysts though and we'll have to trust them to make a good decision.
But what is a good decision in the case of a virus that has a strong track record for killing a significant percentage of those it infects as well as causing serious harm in many of those who do survive it? I don't know since I'm not an infectious disease doctor. However, I do know about the mechanism of action this drug has and how it attacks the virus. Thus, the title of this thread.